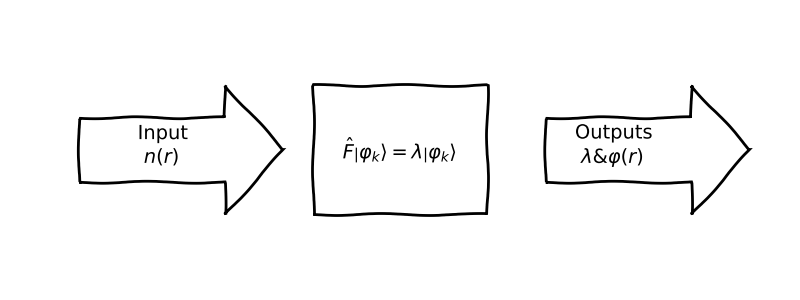
Bottom-up synthesis of Density Functional Theory (DFT)
From the Born-Oppenheimer approximation to the Kohn & Sham equation.Disclaimer
The content of this article is based on various internet resources1. Furthermore, I have attempted to provide a simplified approach to the re-derivation of the equations. Therefore, there may be potential hiccups and mathematicals glichs in some simplifications. Here, I apologize in advance for any mistakes. However, I would welcome any corrections from you.
1:
There was a Turkish website from Istanbul University with in-depth derivations, if I remember correctly. Unfortunately, I stupidly forgot to keep track of the URL. Thousand apologies for that 😅.
“Today we know less than 1% of known compounds in the world; less than 1% of the materials properties.”Gerbrand Ceder @ The Material Desing Revolution the World Economic Forum (2014, Davoz).
A mathematical model about a sufficiently large set of natural phenomena is called an “ab initio model ” or a “first principles model” if the starting point of the model can not be further reduced to more fundamental concepts (at least not within the language of the considered natural phenomena).
By Pr. Stefaan Cottenier @ Ghent University.
Furhtermore, the main idea is to tame the following beast:
The right-hand side of \((2)\) are respectively: electron and nucleuskinetic energy, electron-electron and nuclei-nuclieCoulomb interaction and the electron-nuclieCoulomb interaction.
The basic idea is the following: ”We consider the nuclei as stationary objects, so we freeze the nuclear positions.”. We say that this approximation is adiabatic (We move from \(4(Ne + Nn) \longrightarrow 4Ne\)).
💡 The main idea:For all the possible sets of the wavefunctions that can describe our system, we search for the one associated to the ground state.
With the following restrictions:
Just the anti-symmetric wavefunctions;
Use Salter determinant;
The energy minimization principal.
⚠️ At the end this is an approximation (work well for chemistry topics, not for solids).
The first step is visualizing the terms in the reduced Schrödinger equation after \(\textbf{BO}\) approximation. After all, we end up with:
Notice that we omitted the constant term, the reason will be more obvious in the following derivations. The Hartree-Fock method is a variational, wavefunction-based approach. Although it is a many-body technique, the approach followed is that of a single-particle picture, i.e. the electrons are considered as occupying single-particle orbitals making up the wavefunction. Each electron feels the presence of the other electrons indirectly through an effective potential. Each orbital, thus, is affected by the presence of electrons in other orbitals. Now to build our wavefunction, instinctively we write our function as the product of the orbitals occupied by the electrons, and for simplicity we drop the spin degrees of freedom:
❗ But wait, this expression violate the first restriction mentioned above ! We need another tools that respect the anti-symmetric feature of the wavefunction.
The trick that works with me is to write down \(\textcolor{green}{first}\) the orbitals that the Hamiltonian is mainly! applied on. Then we write the remaining terms as single \(bra-ket\) products :
Here we have one permutation operators \(P\) acting on the order of the set of orbitals \(\{1, 2, \dots, n, \dots, m, \dots, N\}\). It is not obvious, but clearly we are expecting something to pop-up for sure! And the idea is to separate all the \(even\) and the \(odd\) permutations in the previous set:
The \(F\) stand for \(Fock\) operator in the last equation. Now we will consider small change in our wavefunction basis (\(\varphi \longrightarrow \varphi + \delta \varphi\)). We first treat the the term with the \(Langrange\) multipliers:
Where \(C.C\) stand for \(Complex\ Conjugate\). Then we work with second term of \(Hartree-Fock\) energy (\(E_{HF}\)), step by step we will start by the electrons coulomb interaction:
Since \(\{r_i;r_j\}\) are \(dummy\) variables, the integral value doesn’t change if we swap the indices. Also, the operator \(\hat{h}_2\) is invariant by changing the order (\(\hat{h}_2\) represent a coulomb interaction ):
It is not obvious, and the following argument that I rise here may be naive! but the factor of half disappear. Why? We can justify it by the interchanging the indices \(i\ \&\ j\) order, or we say at some point of the summation; the states are reached twice! At this point, it is more proper and urgent! to write the last equation in its integral form:
Now we have our final \(action\) expression (\(F\)) of our system, we take the derivative of \(F\) with respect to some \(k^{th}\) bra (\(\bra{\delta\varphi_k} \equiv \varphi^*_k\)). Notice that \(\frac{\delta C.C}{\delta\varphi} = 0\) (the reason for choosing the \(bra\) over the \(ket\) will be obvious at the end!).
The idea of factoring the \(ket\) is just to re-write our expression in Schrodinger-equation like form, and it’s not rigorous from the mathematical point of view! Hence, Factoring out the \(\ket{\varphi_k}\) gives:
This is the why we choose \(bra\) over \(ket\), to end-up with system of Schrodinger-like equations! for which \(\hat{J}_i\) is an integral operator stand for the classical interaction of an electron distributions given by \(|\varphi_j|^2\) and is called the \(\textbf{direct\ term}\). While \(\hat{K}_j\) called the \(\textbf{exchange operator}\), has no classical analogue and it is a direct result of the antisymmetry property of the wavefunction. The Fock operator is given by:
In this section we use \(Koopman's\ theorem\) to give an insight interpretation to the eigenenergies \(\varepsilon_k$\) we find in the previous equation. We will consider two system with (N-1) and N electrons respectively. For which the system of \(N-1\) electron is missing one electron on some orbital “n”. Also, beside omitting the spin orbitals degrees of freedom, we postulate that the orbitals are unchanged during this process.
We start by writing the HF energy of both system:
Notice that for first line we do not have the “n” orbital in our sum. Now, we evaluate the difference \(E_{HF}^{N-1}-E_{HF}^{N}\) term by term through the full expansion of both summations:
Since the indices \(k\) and \(j\) are irrelevant (\(k\equiv j\)), and by make use of the identity \(\bra{f_1f_2}\hat{A}\ket{f_3f_4} = \bra{f_2f_1}\hat{A}\ket{f_4f_3}\), we get:
One of the attempt to solve the \(\textbf{HF}\) equation was done in 1951 by \(\textbf{Roothaan}\). Clemens C. J. Roothaan was a Dutch physicist and chemist known for his development of the self-consistent field theory of molecular structure2. For a quick read brows the Roothaan equation
.
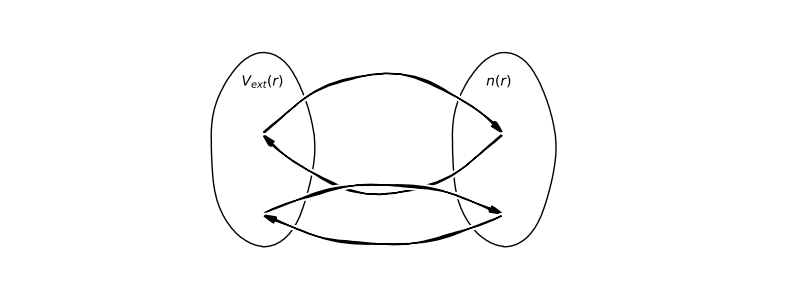
In this section we will rewrite the Hamiltonian as function of the electron density. Again we will treat the Hamiltonian as a spin-less problem. This is the core idea behind density dunctional theory where the function we are interested in take another function and return an output a.k.a number (\(f \longrightarrow I[f]\)).
But first we need to write the later entity \(n(r)\). So to start we define \(n(r_1)\) the electron density of finding an electron at position \(r_1\) while the rest of electrons set are at positions \(\{r_2,r_3, \dots, r_N\}\) as following:
First thing to notice is that we can write the product of the \(bra-ket\) function as the norm squared \(|\Psi(r_1, r_2, \dots, r_N)|^2\) (the operator \(\hat{n}(r_1)\) does not acts on the wavefunction) which yields:
By following the same reasoning the electron density of many-body system \(n(r)\) is given by the sum of all individual electrons densities(the definition \(n(r)= \sum_i^N n(r_i)\), is justified by seeing the operator of \(\hat{n}(r)\) as a linear one):
Since the propriety of the Norm squared of the wavefunction does not change with its arguments shuffled(\(|\Psi(r_1, \dots r_i,r_j,\dots, r_N)|^2 = |\Psi(r_1, \dots r_j,r_i,\dots, r_N)|^2\)). It is obvious that we end-up having \(N\) equal terms, because of the indistinguishable nature of electrons:
Previously we had established the electrons-ions interaction, now we will introduce the electrons density \(n(r)\). We start as always by taking the expectation values of the later coulomb interaction:
The factor of each individual coulomb interaction integral is the same! Beside the same trick of shuffling the arguments and changing re-arranging the indices, we use the dummy variables \(r_i \longrightarrow r\) we rewrite:
Using the same manner that we did in the last section we will introduce the electron density. Note that it is not quite the same quantity \(n(r)\)! Within the electrons-electrons interaction. And the starting point is always the avearage value:
Basically the integral over the \(\mathbb{R}^{3(N-2)}\) space is the same, and we have \((N-1)\) term. And again the shuffling propriety of the wave function:
Yet, we did not write our Energy as a functional of the density \(n(r)\). We still have the Kinetic term which is hard to deal with since we have the operator \(\nabla^2\) that act on the wavefunction (\(\Psi\)). In order to tackle the kinetic energy, we make one of the key assumptions of density functional theory. We assume that the density can be written as the sum norm squares of a collection of single-particle orbitals:
These orbitals are called \(\textbf{Kohn-Sham orbitals}\) and they are initially completely unspecified in much the same way as in the orbitals in the Slater determinant in the \(\textbf{Hartree-Fock}\) formalism. The above form cannot really be considered an
approximation. It simply says that instead of the full many-particle system we consider an auxiliary system of single-particle orbitals that have the same ground state density as the real system. Hence the Kinetic energy can be written in term to the KS orbitals to a correction term; as following:
In the following we will refer to the paper of \(\textsc{Kohn and Sham}\)3. As it was reported in the later paper, the ground state energy has the following form:
Let assume that exist two different external potential \(V_{ext}^{(1)}(r)\) and \(V_{ext}^{(2)}(r)\) that gives rise to the same electrons density for non-degenerate ground state \(n(r)\). Hence, we have two wavefunction for two Hamiltonian:
We will try to re-derive the \(\textbf{KS}\) equation by following the same procedures used in \(\textbf{HF}\) approach. The idea is as simple as the minimization over the wavefunction! But an approximation is needed. As we discussed previously we already write the Energy as functional of the density (i.e: \(E[n(r)]\)); but it was shown that the kinetic energy raise a problem, that it can not be written in terms of the density \(n(r)\), so we consider the \(\textbf{KS}\) orbitals that builds the density from ground i.e: \(n(r)=\sum_i^N |\varphi(r)|^2\), hence the kinetic term can be minimized through the set of these orbitals! that we postulate theirs orthogonality constraints:
In this section we will see how we can solve \(\textbf{KS-equation}\), but first we will write down the matrix formula equivalent to the Schrodinger-like equation; using the Planewave expansion. Without further ado, we start by defining our wavefunction (i.e: the \(\textbf{KS-orbitals}\)) as following:
Where, \(\Omega\) is the volume of the real space where we are working in, and \(c_{i,q}\) are the Fourier coefficients of the wavefunction. Note, that we are moving to the reciprocal space \({\overrightarrow{q}}\) instead of the real one, which will make \(\textbf{Fourier transform}\) the pillars of this work! For our Hamiltonian that is described by \(\textbf{KS}\) equation will be rewritten as follwing:
Where, \(\hat{T}_{s}\) is the single-particle kinetic energy and \(\hat{V}_{eff}\). Where \(\hat{V}_{eff} = \hat{\Theta} + \hat{V}_{xc}\). Is the effective potential which contains the external, \(\textbf{Hartree}\) (i.e: \(\hat{\Theta}\)) and \(\textbf{exchange-correlation}\) (i.e: \(\hat{V}_{xc}\)) parts. So far we can write the wavefunction in the basis set of \(\{\overrightarrow{q}\}\) as following:
where \(\underline{\underline{H}}\) is the Hamiltonian in matrix representation and \(\underline{\underline{C}}\) is a vector of coefficients. Now, we will work out the kinetic energy term:
In the planewave representation, the kinetic energy term assumes an extremely simple, diagonal form. Now we proceed with effective potential, since \(V_{eff}\) has the periodicity of the lattice and therefore the only allowed Fourier components are those with
the wavevectors in the reciprocal space of the crystal:
By make use of the change in variable, by make \(\overrightarrow{q} = \overrightarrow{k}+\overrightarrow{G}_m\) and \(\overrightarrow{q'} = \overrightarrow{k}+\overrightarrow{G}_m'\), so the wave vectors (\(q\ \&\ q'\)) differ by a reciprocal lattice vector. which brings the Schrodinger-equation like to the \(k\) space:
In summary, we reduced the problem into the First \(\textbf{Brillouin zone}\) (The primitive cell in the reciprocal space), thanks to \(\textbf{Bloch's theorem}\). Now it is important to see how the \(V_{eff}\) can be written in the \textbf{Fourier Space}. For the sake of notation we denote \(n(\overrightarrow{r})\) the real electron density and \(\rho(\overrightarrow{G})\) it conjugate in the reciprocal space.
5.1 The electron-electron interaction (Hartee Energy term)
We write : \(\overrightarrow{u} = \overrightarrow{r}-\overrightarrow{r'} \longleftrightarrow d\overrightarrow{u} = \overrightarrow{r}\), and in the first exponential we add and subtract \(\overrightarrow{r'}\):
We separate the independent integral and write \(\underset{\mathbb{R}^3}{\int d\overrightarrow{u}} =2\pi\int_{0}^{\infty}\int_{0}^{\pi} u^2 du sin(\theta) d\theta\):
\[
\begin{align*}
\pi\sum_{\overrightarrow{G'}, \overrightarrow{G}} \rho(\overrightarrow{G'})\rho(\overrightarrow{G}) \underbrace{\int e^{j\overrightarrow{r'}\cdot\Big(\overrightarrow{G'}+\overrightarrow{G}\Big)} d\overrightarrow{r'} }_{\Omega_{cell}\times \delta(\overrightarrow{G'}-\overrightarrow{G}) }\int_{0}^{\infty} \Big[\int_{0}^{\pi} \underbrace{e^{j G\cdot u cos(\theta)} u sin(\theta) d\theta}_{\equiv - \frac{e^{jx}}{G}dx \ |\ x = G\cdot u cos(\theta)\ |\ 1 \langle x \langle -1}\Big]d u\\
\iint v_{e-e}(\overrightarrow{r'}, \overrightarrow{r})n(\overrightarrow{r})n(\overrightarrow{r'})d\overrightarrow{r}d\overrightarrow{r'} = \pi\Omega_{cell}\sum_{\overrightarrow{G'}, \overrightarrow{G}} \rho(\overrightarrow{G'})\rho(\overrightarrow{G})\times \delta(\overrightarrow{G'}-\overrightarrow{G}) \int_{0}^{\infty} \underbrace{\frac{e^{jG\cdot u}}{jG}|_{-1}^{1}}_{2\times \frac{sin(Gu)}{G}} du
\end{align*}
\]
The last integral factor it is not a proper integral!
A further simplification: \(G = -\nu G'\), because the density is a physical measurable quantity, \(\mathcal{F}\{\Phi\}(\omega)=\mathcal{F}\{\Phi\}(-\omega)\), where \(\mathcal{F}\) is Fourier transform and \(\Phi\) physical entity i.e the negative and the positive frequency components are equal [see chapter 8 of Aschroft and Mermin’s “Solid State Physics”].
And by considering the propriety : \(\rho(\overrightarrow{G}) = \rho(\overrightarrow{G'})\)
As we have seen before that the last contribution of the effective potential in \(\textbf{KS}\) Hamiltonian is due to the change of exchange-correlation energy function over a small variation in the electron density (i.e: \(V_{xc}(\overrightarrow{r}) = \frac{\delta E[n(\overrightarrow{r})]}{\delta n(\overrightarrow{r})}\)). We will use the expression given by Walter Kohn et al. 3 for the later term, and since we can exchange the derivation of a function over the integral like the case with regular functions, note that we could derive first and perform the Fourier decomposition, and we ends up by having two terms: